Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA) are rare neurodegenerative diseases, which cause progressive, proximal-to-distal muscular weakness leading to loss of ambulation and motor function. Worldwide, they are the leading cause of neuromuscular disorders affecting children, which has led to an active research field to discover new DMD and SMA therapies.

In February 2018, the US Food and Drug Administration (FDA) issued a new guidance on DMD [1], which outlines the challenges of endpoint definition to demonstrate treatment benefit. Since then, there have been 23 pharmaceutical studies from 17 sponsors studying 17 interventions for DMD, which notably include regulatory drug approvals and fast track (see Table).
Table: Approval, Fast Track and News
| Date | Heading | Notes | Link |
| Feb 25, 2021 | The FDA approves targeted treatment for rare DMD mutation | The FDA approved Amondys 45 (casimersen) injection for patients with confirmed mutation of the DMD gene that is amendable to exon 45 skipping. Amondys 45 increases dystrophin (protein) production in skeletal muscles. It is the first FDA-approved targeted treatment for patients with this type of mutation, which is approximately 8% of DMD patients. Clinical benefit, including improved motor function, has not been established. | https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0 |
| Jan 8, 2021 | First participant dosed in Pfizer’s Phase 3 gene therapy trial for DMD | Pfizer announced that the first participant in the CIFFREO trial studying the potential gene therapy of PF-06939926 has been dosed. | https://www.musculardystrophyuk.org/news/breaking-research-news/ |
| Dec 3, 2020 | Genethon receives go ahead for DMD gene therapy trial in France | Genethon received permission to start trial assessing GNT 004. | https://www.musculardystrophyuk.org/news/breaking-research-news/ |
| Oct 20, 2020 | The FDA grants fast track designation for ASP03067/MA-0211, a selective PPARS Modulator being developed for the treatment of Primary Mitochondrial Myopathies | Astellas Pharma announced that the FDA has granted fast track status for the development of ASP0367 for treating primary mitochondrial myopathies (PMM), which causes reduced muscle function, fatigue, and muscle atrophy. | https://www.astellas.com/us/news/5231 |
| Dec 12, 2019 | The FDA grants accelerated approval to first targeted treatment for rare DMD mutation | The FDA approved Vyondys 53 (Golodirsen) to treat DMD patients who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping, about 8% of DMD patients. Clinical benefit, including improved motor function, has not been established. | https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation |
| Feb 9, 2017 | The FDA approves drug to treat DMD | The FDA approved Emflaza (deflazacort) to treat DMD. Emflaza is a corticosteroid that decreases inflammation and reduces the activity of the immune system. Corticosteroids are commonly used to treat DMD across the world, but this is the first FDA approval of any corticosteroid to treat DMD in the USA. | https://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy |
Selecting the right endpoint for neurodegenerative clinical trials is not trivial. One complication stems from the heterogeneity of disease manifestation in DMD and SMA. This imposes the necessity for multiple endpoint specification to enable the capture of different severity and stages. The resultant multiplicity of tests requires careful management. [2] Another challenge is finding measures of function that cover the spectrum of disease severity, thereby avoiding floor/ceiling effects, which will jeopardize demonstration of treatment benefit. However, these are not easy to find.
For example, the current FDA Guidance [1] flags the limitations of the ubiquitous 6-minute (or shorter 2-minute) walk test. This is a commonly used mobility Performance Outcome [PerfO], which consists of measuring the distance an individual is able to walk over a total of six minutes on a hard, flat surface, as it can show “a floor effect of losing ambulation in older patients with more advanced disease” (p.7). In addition, the clinical meaning of this type of simple, objective timed in-laboratory/clinic test is difficult to interpret. The FDA also acknowledges that Patient-Reported Outcomes can provide useful supportive endpoint data to document the “clinical meaningfulness” of changes of “relatively small magnitude” in “objective” endpoints and “to contribute to assessments of benefit and risk” (p.7) [1].
Thus, sponsors of DMD clinical trials are now invited by the FDA to propose innovative approaches to study endpoints in DMD to “validly and reliably assess patients with a wide spectrum of symptoms and disease stages”. The kind of innovation, which we support and described further elsewhere [3].
Did you know…
Since 2013, the North Star Ambulatory Assessment (NSAA) [4] [5] has been used as an outcome measure in 47 DMD clinical studies. The NSAA is a 17-item motor performance PerfO, consisting of clinically meaningful tasks (items) related to ambulatory performance in DMD. Importantly, the current FDA Guidance supports the inclusion of “the North Star Ambulatory Assessment (NSAA) [as it] can provide a useful measure of gross motor function” (p.7) [1].
One of the lead NSAA researchers is Dr Anna Mayhew, who is a consultant research physiotherapist at the John Walton Muscular Dystrophy Research Centre, Newcastle University, UK. Anna is also a key member of our Scientific Advisory Board, and plays a key role directly advising us on our paediatric neurodegenerative clinical trials research.
Previously, Anna collaborated with our own Chief Scientific Officer, Dr Stefan Cano, to analyse the NSAA using Rasch Measurement Theory (RMT) to improve its interpretability in clinical research.
In an initial study, Anna and Stefan psychometrically analysed cross-sectional NSAA data from ambulant (n=191; mean age 8 [SD 2] years) with a confirmed diagnosis of DMD [6]. Part of this analyses included an assessment of clinical validity. This involved a comparison of the clinical hierarchy of items as rated by five expert neuromuscular physiotherapists to that generated by RMT analysis. The correlation between the ranking of the items of the two methods was very high (rho=0.80). Thus, tasks (items) including stand, walk, gets to sitting, and standing on one leg were defined as easier, and run, jump, and hop, rated more difficult by both experts and RMT analysis.
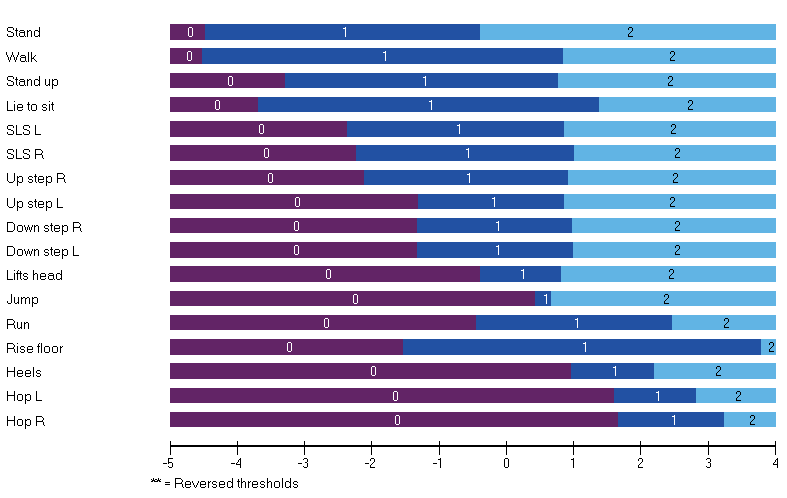
This clinical validity information proved invaluable to interpret the findings of a follow-up study, which focussed on longitudinal data (n=198 boys with DMD on different corticosteroid regimes) [7]. Analyses of the NSAA’s ability to detect clinical differences/change included mean score changes (using Rasch-transformed data) between adjacent pairs of age groups, pairwise squared t-values from paired samples t-tests, effect size and MID calculations. Difference between the two corticosteroid regime cohorts, change scores over time, and MIDs could all be interpreted through the RMT generated clinical hierarchy of items, which provides a readily meaningful frame of reference rooted in the scale content.
The success of these studies subsequently led to the involvement of Anna and Stefan in a large international multi-institutional study to address the issues in the use of functional rating scales measures as study endpoints in pivotal efficacy studies. The Rasch Task Force of the International Coordinating Committee for SMA Clinical Trials (ICC) included representatives of all the study groups involved in developing the most widely used functional rating scales in SMA and resulted in recommendations for future development and improvement for use as clinical trial outcome measures [8]. This led to the improvement of legacy instruments [9] and development of new measures [10].
For further information about how Modus Outcomes can support the use of patient-centered outcome measures in current and future DMD and SMA clinical trials, please contact Melody Bauer.
Further Reading
[1] US Food and Drug Administration. (2018). Duchenne muscular dystrophy and related dystrophinopathies: Developing drugs for treatment. Guidance for industry. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Silver Spring, MD.
[2] US Food and Drug Administration. (2017). Multiple Endpoints in Clinical Trials—Guidance for Industry. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Silver Spring, MD.
[3] Morel T., Cano S.J. Measuring what matters to rare disease patients–reflections on the work by the IRDiRC taskforce on patient-centered outcome measures. Orphanet Journal of Rare Diseases. 2017;12:171.
[4] Scott E., Eagle M., Main M., Sheehan J. The North Star Ambulatory Assessment. Poster presented at the Annual Meeting of the British Paediatric Neurology Association, 2006. Dev Med Child Neurol, 2006.
[5] Mazzone E.S., Messina S., Vasco G., Main M., Eagle M., D’Amico A., et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscular Disorders. 2009; 19(7):458-461.
[6] Mayhew A., Cano S., Scott E., Eagle M., Bushby K., Muntoni, F. Moving towards meaningful measurement: Rasch analysis of the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Developmental Medicine & Child Neurology. 2001; 53(6):535-542.
[7] Mayhew A.G., Cano S.J., Scott E., Eagle M., Bushby K., Manzur, A., Muntoni, F., North Star Clinical Network for Neuromuscular Disease. (2013). Detecting meaningful change using the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Developmental Medicine & Child Neurology. 2013; 55(11):1046-1052.
[8] Cano S.J., Mayhew A., Glanzman A.M., Krosschell K.J., Swoboda K.J., Main M., et al. Rasch analysis of clinical outcome measures in spinal muscular atrophy. Muscle & nerve, 2014; 49(3):422-430.
[9] Pera M.C., Coratti G., Forcina N., Mazzone E.S., Scoto M., Montes J., et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC neurology. 2017; 17(1):39.
[10] Klingels K., Mayhew A.G., Mazzone E.S., Duong T., Decostre V., Werlauff U., et al. Development of a patient‐reported outcome measure for upper limb function in Duchenne muscular dystrophy: DMD Upper Limb PROM. Developmental Medicine & Child Neurology. 2017; 59(2):224-231.